Stefano Gasparini, Martina Bonifazi, Lina Zuccatosta
Dipartimento di Scienze Biologiche e Sanità Pubblica
Università Politecnica delle Marche
SOD di Pneumologia
AOU “Ospedali Riuniti”, Ancona
Claudia Duranti, Maria Agnese Latini
Medici in Formazione
Scuola di Specializzazione in Malattie dell’Apparato Respiratorio
Università Politecnica delle Marche
Scenario clinico
Un uomo di 47 anni giungeva alla nostra attenzione per la comparsa di dispnea da sforzo da circa tre mesi associata al riscontro emogasanalitico di insufficienza respiratoria parziale. Dall’anamnesi emergevano abitudine tabagica, psoriasi cutanea con localizzazione articolare non in trattamento al momento dell’osservazione, nessuna esposizione ambientale o professionale a nocivi ed agenti organici, non familiarità per patologie polmonari.
Gli esami ematici all’ingresso in reparto risultavano nei limiti di norma; in particolare l’assenza di alterazioni degli indici di funzionalità miocardica rendeva meno probabile una genesi congestizia, ed allo stesso modo, la normalità dell’emoglobina escludeva la presenza di una anemia sottostante tale da giustificare il sintomo.
Le prove di funzionalità respiratoria mostravano un deficit ventilatorio di tipo misto di grado discretamente elevato con moderata riduzione della DLCO.
Al test del cammino dei sei minuti percorreva 500 m con una saturazione minima pari al 92%.
In considerazione del dato anamnestico di artrite psoriasica e della componente restrittiva alle prove funzionali si eseguiva lo studio della sierologia immunitaria che risultava nei limiti di norma.
A completamento diagnostico, considerando le alterazioni alle prove di funzionalità respiratoria e l’insufficienza respiratoria parziale, il paziente veniva sottoposto a HRTC che evidenziava un quadro caratterizzato da segni di enfisema centrolobulare parasettale in associazione ad ispessimento dell’interstizio settale interlobulare e perilobulare con modesta distorsione dell’architettura parenchimale, aree di ground glass diffuse, e minimo versamento pericardico. Alle scansioni passanti per l’addome (Fig.1) si documentava addensamento del tessuto perirenale bilateralmente.
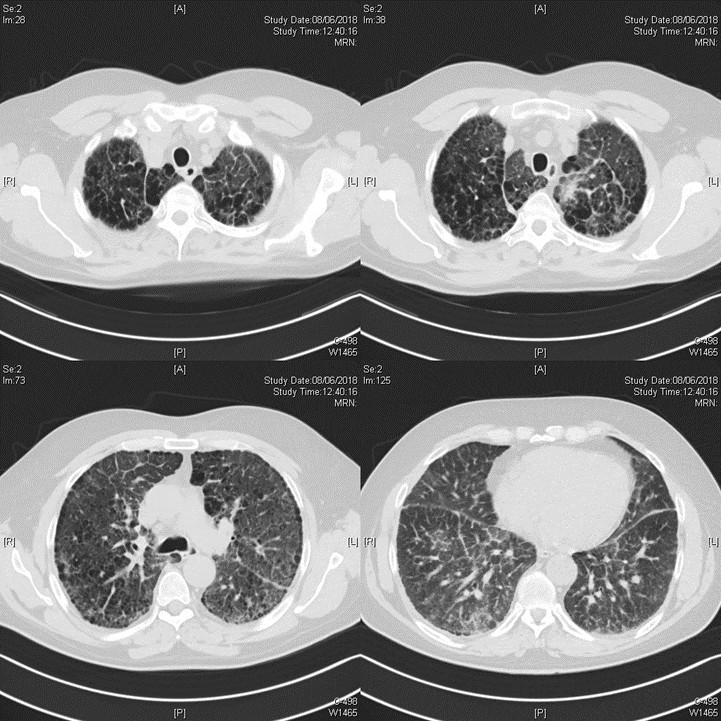
Figura 1- quadro polmonare caratterizzato da ispessimento liscio dell’interstizio settale, aree di ground glass ed ispessimento pleurico
Alla luce dei dati anamnestici e strumentali si configurava, quindi, il seguente problema clinico: insufficienza respiratoria parziale e deficit ventilatorio misto associati a reperto radiologico di enfisema e ispessimento dell’interstizio in soggetto fumatore affetto da artrite psoriasica.
Sulla base dei dati raccolti era possibile formulare le seguenti ipotesi diagnostiche:
– Enfisema polmonare centrolobulare e parassettale con componente di edema polmonare acuto o linfangite carcinomatosa (infiltrazione dei vasi linfatici polmonari da parte di cellule neoplastiche. Si associa più frequentemente a tumori primitivi polmonari, mammari, pancreatici, gastricocolici e prostatici);
– Erdheim-Chester disease (Rara forma di istiocitosi a cellule non Langerhans caratterizzata dall’infiltrazione sistemica da parte di istiociti CD68+ e CD1a- privi di granuli di Birbeck. La diagnosi in età adulta la contraddistingue dalle altre forme di istiocitosi non Langerhans. La manifestazione clinica è eterogenea in relazione alla variabilità degli organi coinvolti. Il coinvolgimento osseo avviene nel 95% dei casi. Le manifestazioni extra ossee interessano più frequentemente il retroperitoneo in particolare a livello perirenale, il sistema nervoso centrale, il torace come localizzazione miocardica, pericardica, polmonare, mediastinica, pleurica, vascolare);
– Malattia veno-occlusiva (Malattia rara caratterizzata da restringimento delle venule polmonari causata dalla fibrosi dell’intima. Si associa a ipertensione polmonare postcapillare);
– Edema polmonare acuto interstiziale (accumulo interstiziale di liquidi, prevalentemente in sede peribroncovascolare e a livello dei setti interlobulari. L’assenza di distorsione del parenchima polmonare e l’ispessimento liscio prevalentemente lineare dei setti interlobulari possono aiutare a differenziare un edema interstiziale cardiogeno da una pneumopatia infiltrativa diffusa).
La presentazione clinica e gli accertamenti effettuati orientavano maggiormente verso una possibile malattia di Erdheim Chester. Venivano dunque formulati alcuni quesiti.
Quesito diagnostico: sono sufficienti i dati clinici e radiologici per porre diagnosi di malattia di Erdheim Chester? Quali esami consentono di porre la diagnosi?
La diagnosi di malattia di Erdheim Chester (ECD) viene formulata identificando le caratteristiche lesioni istopatologiche nel giusto contesto clinico e radiologico: presenza di infiltrati di istiociti schiumosi o carichi di lipidi circondati da flogosi e/o fibrosi, molto spesso associati alla presenza di cellule giganti multinucleate. La diagnosi di Erdheim Chester, per una corretta formulazione, necessita quindi di un riscontro istologico a supporto di un suggestivo contesto clinico.
Pur condividendo alcune caratteristiche con l’istiocitosi a cellule di Langherans (LCH), la malattia di Erdheim Chester rappresenta un’entità nosologica distinta, dovuta a proliferazione clonale di istiociti caratterizzata da specifici elementi morfologici ed un definito profilo immunoistochimico (Tabella I): gli istiociti schiumosi della malattia di Erdheim-Chester sono caratterizzati da positività per il CD68, il CD163 ed il Fattore XIIIa, come quelli dell’ICL, ma negativi per il CD1a e la langherina (CD207).
| ECD | LCH | |
| Caratteristiche istopatologiche | ||
| CD68 | + | + |
| CD163 | + | + |
| CD1a | − | + |
| CD207 | − | + |
| S100 | − o debolmente + | + |
| Fattore XIIIa | + | − |
| Cellule giganti multinucleate | + | − |
| Altre caratteristiche tipiche delle lesioni istiocitarie | Xantomatosi
Fibrosis |
Granuli di Birbeck in microscopia elettronica |
Tabella I – Caratteristiche istopatologiche della malattia di Erdheim-Chester e dell’istiocitosi a cellule di Langherans (modificata da Diamond et al. Consensus guidelines for the diagnosis and clinical management of ECD).
L’interessamento osseo, presente nella pressochè totalità dei pazienti, è la più comune manifestazione clinica, con tipico coinvolgimento delle diafisi e metafisi degli arti inferiori: le lesioni osteosclerotiche simmetriche e bilaterali risultano evidenziabili mediante esame radiografico, scintigrafia ossea, PET-TC e, in casi selezionati, mediante RMN.
Altro tipico reperto della malattia di Erdheim-Chester è rappresentato dall’hairy kidney, ovvero un’infiltrazione dello spazio retroperitoneale, documentabile alla TC nel 30% circa dei pazienti.
Anche in caso di quadri clinico-radiologici altamente suggestivi per malattia di Erdheim Chester la biopsia è comunque mandatoria soprattutto per valutare lo stato mutazionale del gene BRAF, alla luce delle importanti implicazioni terapeutiche che ne derivano. Infatti, la scoperta della mutazione BRAF-V600E, riportata in letteratura nel 38-68% circa dei casi, è risultata fondamentale da un punto di vista terapeutico, essendo oggi disponibili nuovi farmaci che hanno tale alterazione come target di azione.
A completamento dello studio, venivano eseguite ulteriori indagini sia per identificare possibili target per il prelievo istologico sia anche a scopo stadiativo. In particolare, la TC dell’addome confermava la presenza di addensamento del tessuto perirenale bilateralmente e la radiografia delle ossa lunghe degli arti inferiori documentava la presenza di diffuso osteoaddensamento sclerotico e segni di proliferazione periostale a carico delle regioni metadiafisarie con risparmio delle epifisi (Fig.2 A-B). Infine uno studio ecocardiografico escludeva la presenza di disfunzione cardiaca che potesse giustificare una genesi congestizia del quadro radiologico.
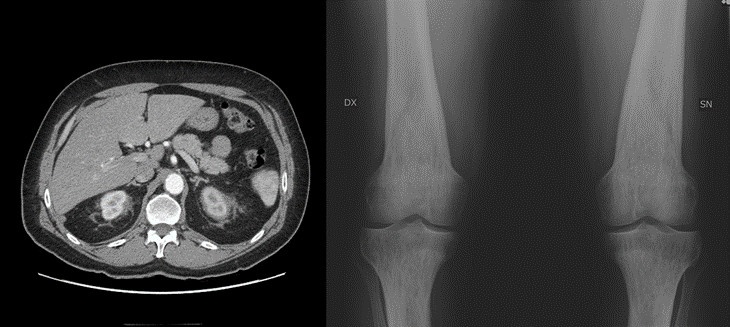
Figura 2.A: – tipico aspetto “hairy kidney” alla TC addome con mezzo di contrasto
Figura 2.B – coinvolgimento osseo, con interessamento della diafisi e metafisi delle ossa degli arti inferiori
Ogni sospetta localizzazione di malattia è un potenziale target per la biopsia, ed ovviamente andrebbe privilegiato il sito il cui approccio presenti il miglior rapporto rischio beneficio, cercando di ottenere campioni adeguati per esame istologico e studio mutazionale. Occorre però tenere presente che non sempre l’esame istologico evidenzia il classico infiltrato di istiociti schiumosi: è possibile infatti incontrare una grande varietà di quadri istologici, inclusa una flogosi aspecifica frammista a fibrosi o anche fibrosi con scarsi istiociti.
Infine è necessario ricordare che nel caso di biopsia ossea per l’interpretazione istopatologica è necessaria la decalcificazione, che può rendere il campione inadeguato per le analisi genetiche: pertanto, è indispensabile ottenere un numero adeguato di prelievi per avere del materiale anche per lo studio del DNA.
Considerato l’interessamento prevalentemente polmonare, il paziente veniva dunque sottoposto a biopsie polmonari transbronchiali mediante criosonda in corrispondenza del lobo polmonare inferiore destro.
La criobiopsia tranbronchiale è una procedura broncoscopica di recente adozione nel work-up diagnostico delle pneumopatie infiltrative diffuse, che si configura come metodica diagnostica alternativa meno invasiva rispetto all’approccio chirurgico, essendo caratterizzata da un miglior profilo rischio-beneficio.
L’esame istologico documentava la presenza di aree di fibrosi moderatamente cellulata da una popolazione infiammatoria mista costituita in parte da elementi istiocitari, accompagnati da alcuni linfociti, scarse plasmacellule e rari granulociti eosinofili.
Le indagini immunoistochimiche evidenziavano la presenza della componente istiocitaria con positività per CD68 e CD163 e negatività per S100 e CD1a. Sebbene focali, tali reperti apparivano compatibili con l’ipotesi diagnostica di malattia di Erdheim-Chester (Fig.3 A-B).
La ricerca della mutazione di BRAF mediante sequenziamento diretto con PCR (mutazione V600E 3 V600K) dava esito negativo.
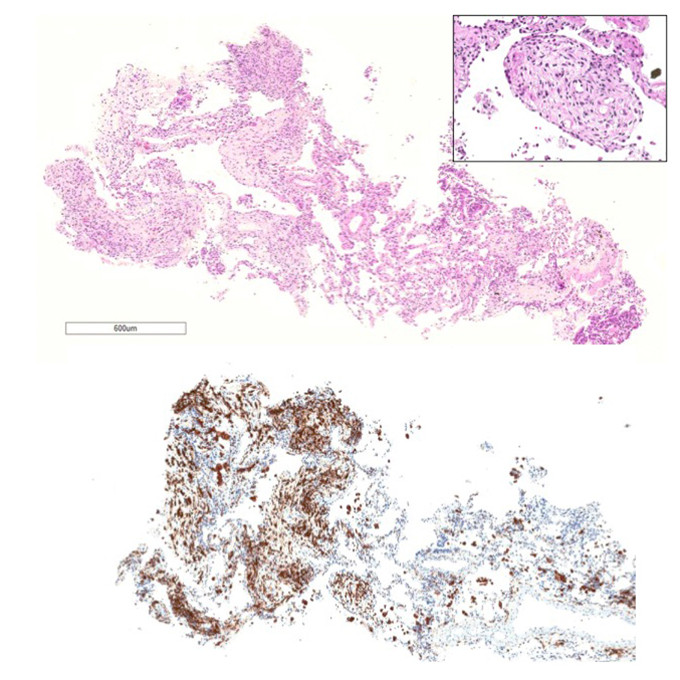
Figura 3.A – Quadro istologico caratterizzato dalla presenza di un infiltrato infiammatorio misto (istiociti, linfociti, scarse plasmacellule, rari granulociti eosinofili).
Figura 3.B – Colorazione immunoistochimica: positività per CD68 della componente istiocitaria.
Quesito terapeutico: in un paziente con malattia di Erdheim Chester è sempre indicata una terapia medica? Quali sono le opzioni terapeutiche disponibili?
Una volta formulata la diagnosi di ECD, il trattamento viene raccomandato in tutti i pazienti sintomatici; negli altri casi è possibile effettuare un attento monitoraggio piuttosto che iniziare immediatamente la cura.
Nel corso degli anni sono stati proposti diversi tipi di trattamento (steroidi, alcaloidi della vinca, antracicline, ciclofosfamide, trapianto autologo di cellule staminali, radioterapia) con risultati piuttosto insoddisfacenti, fino allo sviluppo delle attuali conoscenze sui meccanismi patogenetici. Grazie alla scoperta della presenza di numerose citochine proinfiammatorie sia a livello della lesione che a livello sistemico, responsabili del reclutamento e attivazione degli istiociti, sono stati sviluppati farmaci selettivi in grado di bloccare la cascata delle citochine (anakinra, un antagonista ricombinante del recettore per l’IL1, l’infliximab, un anticorpo monoclonale anti TNF-alfa, il tocilizumab, un anticorpo monoclonale umanizzato contro il recettore dell’interleuchina , il sirolimus, inibitore di mTOR).
L’interferone alfa 2-a riveste un ruolo determinante nella terapia della ECD, essendo associato ad un miglioramento significativo della sopravvivenza globale, sebbene la sua efficacia sia condizionata dalla sede prevalente della malattia.
Successivi studi hanno dimostrato la presenza della mutazione BRAFV600E in gran parte dei pazienti ed alterazioni della via delle MAPK nei soggetti BRAF wyld-type, consentendo lo sviluppo di terapie mirate. Il vemurafenib, inibitore specifico di BRAF, ha significativamente migliorato la prognosi dei pazienti, riducendo la mortalità dal 60% al 20% e inducendo una risposta a sei mesi nel 90% casi.
Sono stati inoltre sviluppati farmaci target che agiscono sulla via RAS-RAF-MEK-ERK, come ad esempio il cobimetinib, un inibitore MEK. Seppur efficaci in termini di miglioramento della sopravvivenza, la loro tossicità richiede una attenta valutazione prima di decidere porre indicazione al trattamento. Per tale motivo è stato recentemente proposto un algoritmo terapeutico che limita l’utilizzo degli inibitori di BRAF e MEK ai soli pazienti con malattia moderata-grave o a coloro che non responsivi ad trattamenti di prima linea.
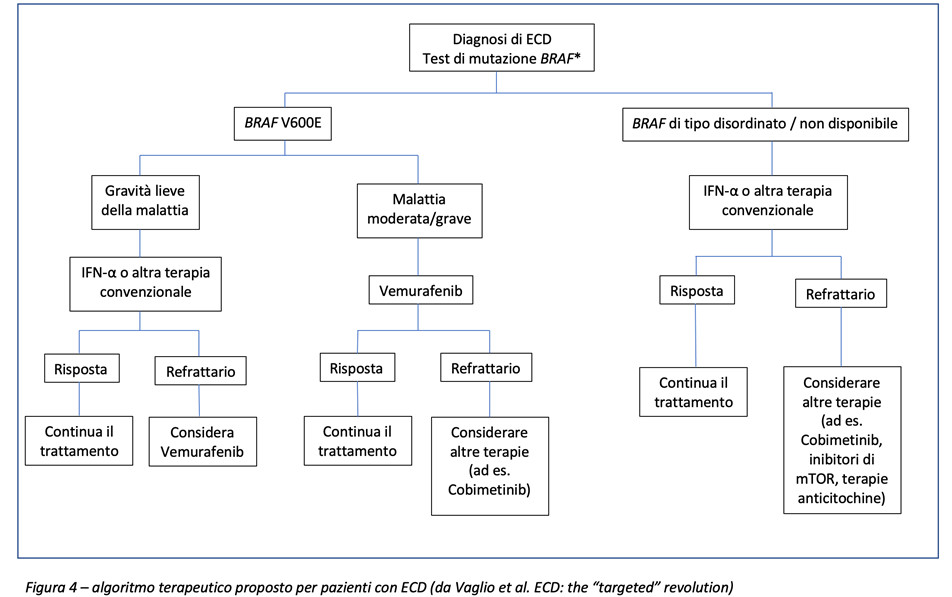
Quesito prognostico: esistono dei fattori prognostici in grado di predire il decorso della malattia?
La malattia di Erdheim-Chester è una patologia caratterizzata da una severa prognosi una percentuale di sopravvivenza a 3 anni inferiore al 50%. Tuttavia, dati più recenti descrivono una sopravvivenza del 68% a 5 anni in pazienti trattati con interferone [5] e si auspica che le nuove terapie target siano in grado di migliorarne ulteriormente la prognosi.
Elementi determinanti nell’evoluzione della patologia sono entità dei sintomi e severità del coinvolgimento d’organo. Nel 2014 è stata proposta una classificazione della malattia di Erdheim-Chester in base alla presenza o assenza di sintomi [1].
| Severità del quadro clinico e prevalente coinvolgimento degli organi sistemici |
| ECD asintomatica o lievemente sintomatica |
| - ECD con prevalente coinvolgimento cutaneo |
| - malattia ossea asintomatica o lievemente sintomatica |
| ECD sintomatica |
| - prevalente coinvolgimento SNC |
| - prevalente coinvolgimento cardiaco |
| - prevalente coinvolgimento retroperitoneale |
| - prevalente coinvolgimento polmonare |
| – prevalente coinvolgimento orbito-craniofacciale |
| - prevalente coinvolgimento neuroendocrino |
| - ECD multisistemica |
Tabella 2 – Classificazione della malattia di Erdheim-Chester (da Diamond et al. Modificata)
Un severo coinvolgimento a carico di qualsiasi distretto costituisce una malattia “ad alto rischio”; per questo motivo il fenotipo clinico di ogni paziente viene meglio caratterizzato dall’organo maggiormente coinvolto, potendo questo influenzare la prognosi.
All’esordio della malattia è raccomandata una valutazione iniziale, volta a definire gli organi potenzialmente coinvolti, l’entità dell’interessamento e la ricerca di possibili localizzazioni asintomatiche. Viene dunque raccomandata l’esecuzione in tutti i pazienti di TC del torace-addome-pelvi, la PET-FDG total body (incluso il cranio e le estremità distali), la RMN dell’encefalo e la RMN cardiaca. Per la valutare l’interessamento del sistema scheletrico è fondamentale l’esecuzione di una radiografia dello scheletro o di una scintigrafia ossea sia nei pazienti sintomatici che in quelli asintomatici.
In aggiunta risultano indispensabili esami di laboratorio per la valutazione di un’eventuale citopenia, di insufficienza renale e di endocrinopatie [1].
Quesito gestionale: una volta posta la diagnosi di malattia di Erdheim Chester è necessario un follow up? Come organizzare il proseguimento dell’iter terapeutico?
La malattia di Edheim Chester è una patologia rara, e pertanto è fondamentale indirizzare il paziente ad un centro di riferimento con un team esperto nella diagnosi e nel trattamento, anche per l’inserimento in trial clinici.
I pochi dati presenti in letteratura suggeriscono una FDG-PET ogni 3-6 mesi dopo l’inizio del trattamento (con aumento dell’intervallo tra gli esami una volta raggiunta una fase di stabilità clinica). In aggiunta a ciò, in caso di interessamento di organo, indagini mirate andrebbero svolte ogni 3 mesi dopo l’inizio del trattamento, ogni 6 mesi una volta stabilizzata la malattia o prima in caso di alterazioni delle condizioni cliniche o degli esami di laboratorio, come la comparsa di una insufficienza renale [1].
Il nostro paziente veniva dunque inviato ad un centro di riferimento per il proseguimento dell’iter diagnostico, completamento della stadiazione e l’avvio di un’ appropriata terapia medica.
Letture raccomandate
- Diamond, E.L., et al., Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood, 2014. 124(4): p. 483-92.
- Munoz, J., et al., Erdheim-Chester disease: characteristics and management. Mayo Clin Proc, 2014. 89(7): p. 985-96.
- Campochiaro, C., et al., Erdheim-Chester disease. Eur J Intern Med, 2015. 26(4): p. 223-9.
- Vaglio, A. and E.L. Diamond, Erdheim-Chester disease: the “targeted” revolution. Blood, 2017. 130(11): p. 1282-1284.
- Cavalli, G., et al., The multifaceted clinical presentations and manifestations of Erdheim-Chester disease: comprehensive review of the literature and of 10 new cases. Ann Rheum Dis, 2013. 72(10): p. 1691-5.