Fabiana Brigante¹, Roberta Mazzucchelli², Andrea Ranghino¹

¹S.D.O. Nefrologia, Dialisi e Trapianto Rene, A.O.U. Ospedali Riuniti, Ancona
²Dipartimento di Anatomia Patologica, Università Politecnica delle Marche, A.O.U. Ospedali Riuniti, Ancona Facoltà di Medicina e Chirurgia, Università Politecnica delle Marche
La presentazione di un caso raro ma esemplificativo di disordine linfoproliferativo associato a crioglobulinemia fornisce agli Autori l’occasione per puntualizzare i meccanismi – immunoflogosi ed ipercoagulabilità – che sono alla base di una forma particolare di glomerulonefrite membrano-proliferativa, quale si può osservare in corso di morbo di Waldenstrom. La ricerca di questo linfoma deve quindi rientrare tra le ipotesi diagnostiche della ne– fropatia descritta. Forme nuove di terapia consentono oggi di migliorare la prognosi a breve e lungo termine dei pazienti che ne sono affetti.
Le glomerulonefriti membranoproliferative altrimenti dette GN mesangiocapillari rappresentano il 7-10% di tutti i casi di GN diagnosticate biopticamente e sono la terza o quarta causa di insufficienza renale cronica in stadio terminale (ESRD) tra le GN primitive. Sono caratterizzate da modalità di presentazione estremamente variabile (dalla microematuria e proteinuria asintomatica, alla proteinuria nefrosica con danno renale acuto fino alle forme di GN rapidamente progressive) e da un decorso che può essere a lenta o rapida evoluzione. Da un punto di vista istopatologico le GNMP si caratterizzano per la presenza di ipercellularità mesangiale, proliferazione endocapillare ed ispessimento della parete capillare attribuibili a deposizione in tale sede di immunoglobuline, fattori del complemento o entrambi che esitano in una sostanziale modifica delle caratteristiche ultrastrutturali del glomerulo.
Sulla base delle caratteristiche documentate alla microscopia elettronica le glomerulonefriti membranoproliferative sono state storicamente classificate in GNMP tipo I, II e III; le forme di tipo I si caratterizzano per la presenza di depositi sub epiteliali, le forme di tipo III per la presenza di depositi subendoteliali e subepiteliali, mentre le forme di tipo II si caratterizzano per la presenza di depositi densi a livello della membrana basale glomerulare (dense deposit disease)¹. Attualmente, le forme di glomerulonefrite membranoproliferative vengono distinte sulla base di criteri eziopatogenetici piuttosto che sulla base di criteri istologici; pertanto le forme di GNMP precedentemente definite di tipo II attualmente comprendono forme complemento-mediate, caratterizzate dalla disregolazione della via alternativa² del complemento. Tale disregolazione è determinata da mutazioni dei geni che codificano per i fattori del complemento o dalla presenza di anticorpi diretti contro le proteine di regolazione del complemento e contro la C3 convertasi con conseguente attivazione incontrollata della via alternativa2 mentre le forme precedentemente identificate come di tipo I e III sono attualmente riconosciute come forme secondarie a deposizione di immunocomplessi (IC) circolanti³.
Le GNMP mediate da immunocomplessi scaturiscono dalla deposizione di questi ultimi a livello glomerulare. Le cause che si associano alla formazione di IC sono riconducibili a paraproteinemia in corso di gammopatie monoclonali, malattie autoimmuni (LES, Sindrome di Sjogren, artrite reumatoide) e infezioni croniche (i.e. HBV, HCV, streptococchi, micobatteri, mycoplasmi, meningococchi e coxielle)⁴.
Dal punto di vista patogenetico, gli IC depositati a livello glomerulare stimolano l’attivazione della via classica del complemento e conseguentemente la deposizione di alcune frazioni dello stesso a livello mesangiale e lungo la parete capillare che culminano nella attivazione della C3 convertasi e termine della cascata complementare. Oltre all’attivazione della cascata del complemento, il danno a carico della parete capillare è sostenuto dall’attivazione dei leucociti e delle citochine che alterano la normale struttura endoteliale e sovvertono la membrana basale glomerulare con conseguente evidenza clinica di ematuria e proteinuria.
Caso clinico
Riportiamo il caso di un uomo di 72 anni, trasferito alla nostra attenzione per quadro di sindrome nefrosica florida con contestuale scarso controllo pressorio e dispnea.
In anamnesi erano presenti ipertensione arteriosa, ipercolesterolemia, ipertrofia prostatica benigna in terapia con alfa-litici e nefrolitiasi. Il paziente riferiva la comparsa di edemi declivi ingravescenti da alcune settimane per i quali è stato ricoverato in un reparto di Medicina di altro Ospedale dove è stato riscontrato un quadro clinico-laboratoristico compatibile con sindrome nefrosica associata a insufficenza renale (creatinina
1.7 mg/dl). In particolare, la perdita proteica urinaria nelle 24 ore (Uprot) era 24.5g/24h. L’immunofissazione sierica è risultata positiva per IgM-k in associazione a proteinuria di Bence Jones con rapporto k/lambda pari a 12.75. Negativa è stata la ricerca dei seguenti anticorpi: ANA, ENA, ANCA, Anti-PLA2R e Anti-GBM. Le frazioni del complemento nel siero erano ridotte. L’ecografia dell’addome mostrava reni nella norma per morfovolumetria con regolare spessore parenchimale, indici di resistenza intraparenchimali pari a 0.8. in assenza di calcoli intrarenali né dilatazione delle vie urinare; non anomalie a carico di fegato, milza, pancreas e aorta addominale. La TC cerebrale mostrava una microimmagine iperdensa a livello della regione nucleare sin, l’Rx scheletro escludeva lesioni osteolitiche mentre l’ecocardiogramma mostrava una iperecogenicità del setto interventricolare. Pertanto, alla luce dei reperti ematologici veniva eseguita biopsia osteomidollare con riscontro di una componente plasmacellulare monoclonale pari al 15%. Quindi, il paziente veniva traferito alla nostra attenzione per approfondimento diagnostico nel sospetto di patologia renale correlata alla paraproteinemia.
All’ingresso in Reparto il paziente presentava ortopnea, SatO2 96% in aria ambiente, PA 180/100 mmHg, peso corporeo 95.7 Kg. Obiettivamente erano presenti edemi colonnari improntabili e simmetrici associati a edema scrotale. Al torace presenza di MV aspro su tutto l’ambito con ronchi diffusi bilateralmente. Non rilievi obiettivi cardiaci e addominali di significato clinico. Si confermava proteinuria in range nefrosico (6 g/24h), alterazione della funzione renale con creatinina 2.2 mg/dl, protidemia totale 4.5 g/dl, albumina 2.4 g/ dl, crasi ematica nei limiti. Veniva confermato severo consumo complementare C3 (58 mg/dl) e C4 (4 mg/dl) con fattore reumatoide sensibilmente elevato (2670 UI/ml). Dato il severo quadro di ritenzione idrica e lo scarso controllo pressorio che controindicavano inizialmente la biopsia renale, si procedeva all’esecuzione di biopsia del grasso periombelicale che escludeva la presenza di amiloide. In considerazione della comparsa durante la degenza di petecchie agli arti inferiori si procedeva a richiedere la ricerca di crioglobuline nel siero risultata positiva con criocrito pari al 5%. Pertanto, si incrementava la terapia steroidea in atto con conseguente miglioramento del quadro cutaneo e della funzione renale (creatinina da 2.8 mg/dl a 2 mg/ dl). Dopo adeguata rimodulazione della terapia antipertensiva ed utilizzo di politerapia diuretica in associazione ad infusione di albumina e.v. che rendevano possibile un calo ponderale di circa 15 Kg ed un soddisfacente controllo pressorio, si procedeva all’esecuzione di biopsia renale percutanea eco-guidata in real-time al polo inferiore del rene sinistro. L’indagine istologica mostrava un quadro compatibile con glomerulonefrite membrano-proliferativa di tipo 1. In particolare in microscopia ottica: 7 glomeruli di cui 1 scleroialino; i restanti mostravano ipercellularità delle anse capillari e inspessimento diffuso delle membrane basali capillari. L’impregnazione argentica mostrava doppi contorni delle membrane basali capillari e irregolarità del loro contorno esterno. L’epitelio tubulare era ridotto di altezza. Erano presenti rari cilindri ialini basofili endotubulari privi di reazione cellulare periferica. Rari tubuli atrofici. Interstizio focalmente ampliato per fibrosi e flogosi crionica. Le piccole arteriole mostravano iperplasia intimale, (Figura 1). L’immunofluorescenza risultava positiva +++ con pattern granulare lungo la membrana basale capillare glomerulare per IgG, IgM, C3, kappa e lmbda con prevalenza kappa. Negativa la ricerca di amiloide.
Si concludeva quindi per una diagnosi di malattia di Waldenstrom con associata glomerulonefrite crioglobulinemica di tipo I. In accordo con i Colleghi Ematologi si impostava terapia con ciclofosfamide, rituximab e desametasone in accordo a protocollo ematologico CRD. Al momento della dimissione il paziente presentava un quadro di stabilità della funzione renale con valori di creatinina pari a 2 mg/dl; ancora obiettivabili gli edemi declivi seppur in netto calo. Dopo circa una settimana dall’inizio della terapia chemioterapica comparsa di malessere generalizzato associato ad astenia, dolore addominale, netto peggioramento degli edemi declivi e della funzione renale (creatinina 4.1 mg/ dl). Pertanto, il paziente veniva nuovamente ricoverato nel nostro Reparto.
All’ingresso: creatinina 4.7 mg/dl, urea 320 mg/dl, uricemia 7.5 mg/dl, proteinuria fino a 6.2 g/die, protidemia tot 4.1 g/dl, albumina 2.1 g/dl, ipocomplementemia (C3: 66 mg/dl, C4: 2 mg/dl), fattore reumatoide (1610 U/ml). Era inoltre presente leucopenia e neutropenia (GB: 1150/mmc; neutrofili 780/mmc) con necessità di somministrazione di granulochine associata a riattivazione virale del citomegalovirus (CMV-DNA: 14.790 U/ml) per la quale è stata impostata terapia con valganciclovir. In considerazione della progressiva contrazione della diuresi associata alla comparsa di segni di stasi polmonare nonostante terapia diuretica massimale si impostava trattamento emodialitico previa posizionamento di catetere venoso centrale in vena giugulare interna destra. Si assisteva inoltre alla ricomparsa di petecchie agli arti inferiori associate a ulteriore incremento del criocrito sino al 12%. In accordo con i Colleghi Ematologi si interrompeva il trattamento chemioterapiaco e si procedeva ad impostare terapia steroidea con prednisone (1 mg/Kg/die) per os associata a trattamenti di plasmaferesi con tecnica a cascata per complessivamente n. 8 trattamenti. Si è assistito ad un graduale miglioramento del quadro clinico con progressiva ripresa della diuresi, miglioramento della funzione renale e netta riduzione del criocrito < 3%. Alla dimissione creatinina 2,2 mg/dl, Uprot 4 g/24h. In seguito su indicazione Ematologica riprendeva la terapia chemioterapica con ciclofosfamide, rituximab e desametasone per complessivamente due cicli. A distanza di sei mesi il paziente si presentava in ottime condizioni generali, eupnoico, normoidratato e con valori pressori controllati. Netta riduzione della perdita proteica urinaria (Uprot 1.5 g/24 h) associata a ulteriore miglioramento della funzione renale (creatinina di 1.2 mg/dl)
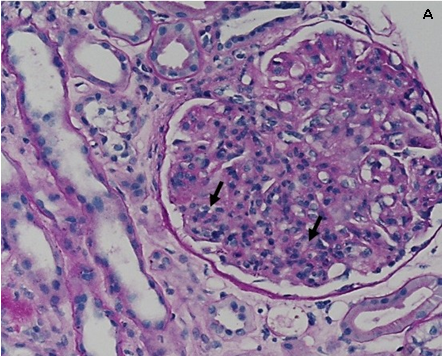
Figura 1: A) ispessimento delle membrane basali glomerulari ed ipercellularità delle anse capillari (frecce nere). (Colorazione PAS)
Discussione
Le gammopatie monoclonali rappresentano condizioni patologiche definite da un’eccessiva secrezione di immunoglobuline secondaria ad un’abnorme proliferazione di un clone di plasmacellule o di linfociti
B. Dal punto di vista patogenetico il danno renale in corso di patologie ematologiche è causato dalla deposizione a livello glomerulare di paraproteine anomale (Ig, componente M) o loro subunità (catene leggere e pesanti, componenti incomplete), a depositi di amiloide e all’infiltrato linfoplasmacellulare interstiziale⁵-⁶. In particolare, sono le complesse interazioni tra le caratteristiche fisico-chimiche delle catene leggere ed il microambiente glomerulo-tubulare (sistemi recettoriali, uromodulina o proteina di Tam Horsfall, fattori di crescita, ecc.) che definiscono la sede di danno renale glomerulare o tubulare e di conseguenza il tipo di lesione istologicamente documentabile⁷.
Tra le malattie linfoproliferative a basso grado di malignità la macroglobulinemia di Waldenstrom (WM), è caratterizzata da una proliferazione clonale di linfociti B e dalla presenza di una componente monoclonale di tipo IgM > 1g/dl associato ad un infiltrato linfoplasmocitico > 10% alla biopsia osteomidollare⁸.
In considerazione della bassa incidenza di WM, pari a solo 1500 casi/ anno negli Stati Uniti, il numero di studi disponibili in letteratura in merito al tipo di danno renale in corso di WM sono nettamente inferiori rispetto a quelli sulla nefropatia in corso mieloma multiplo. Pertanto, in letteratura il quadro istologico maggiormente rappresentato di danno renale in corso di WM è la malattia da depositi monoclonali intracapillari (ICMDD). La ICMDD è caratterizzata da una cospicua deposizione di materiale non amiloide, fortemente PAS positivo, localizzato a livello endoteliale e tale da determinare possibile occlusione del lume capillare. L’etiologia della ICMDD è stato associato alla coesistenza di sindrome da iperviscosità propria della WM nonché alla presenza di crioglobuline circolanti⁹. Negli ultimi anni la miglior gestione terapeutica della patologia linfoproliferativa¹⁰ con conseguente minor concentrazione di IgM monoclonali nel siero ha consentito di dimostrare la presenza di altre alterazioni renali quali le GN amilodosiche, le GNMP associate o meno a crioglobulinemia e le nefriti interstiziali da infiltrati linfo-plasmacellulari10. Recentemente, Higgins L et al11, ha dimostrato in un’ampia casistica di pazienti con WM ed altre malattie linfoproliferative secernenti IgM, la presenza di glomerulopatia amilodosica (33% dei casi), glomerulonefriti (GN) non amiloidosiche (35% dei casi) e nefriti tubulo-interstiziali (14% dei casi). Nello stesso studio, le GN associate a crioglobulinemia rappresentavano la percentuale maggiore tra le GN non amiloidosiche ed erano caratterizzate istologicamente da GN membranoproliferative12-13.
In aggiunta, Neel A et al hanno dimostrato che la maggior parte delle GN non associate a depositi di amiloide mostrano un pattern istologico suggestivo per GNMP tipo 1 con depositi PAS positivi subendoteliali e mesangiali, aspetti di doppio contorno della membrana basale glomerulare e cellule mononucleate nel lume dei capillari glomerulari o di GN crioglobulinemica con depositi subendoteliali di IgM monoclonali e trombi nei capillari glomerulari14-15.
Inoltre, Muria et al16-17 ha dimostrato che è possibile osservare aspetti istopatologici di overlap fra ICMDD e glomerulonefriti membranoproliferative in corso di macroglobulinemia di Waldestrom, soprattutto nelle forme con sovrapposta crioglobulinemia. In tale contesto coesistono lesioni caratterizzate dalla presenza di IgM lambda in sede capillare con immagini di doppio contorno della membrana basale e proliferazione mesangiale.
Altri Autori hanno documentato la presenza di aspetti di microangiopatia trombotica (TMA) nelle biopsie renali di pazienti con macroglobulinemia di Waldestrom che solo in alcuni casi si associa va alla chemioterapia (18) ed in assenza di segni sistemici di danno microangiopatico (i.e. no piastrinopenia, no incremento degli indici di emolisi). Diversamente da quanto apprezzato in corso di ICMDD dove gli aggregati trombotici intracapillari sono costituiti da aggregazioni di IgM monoclonali negativi per fibrina, nei casi descritti di TMA in corso di Waldestrom si è soliti riscontrare trombi endocapillari costituiti da piastrine e fibrina e negativi per IgM. Il meccanismo patogenetico di tali lesioni non è noto tuttavia, l’ipotesi più verosimile si basa sullo stato di iperviscosità che determinerebbe uno shear stress endoteliale con conseguente maggior rischio protrombotico in aggiunta a quello determinato dalle paraproteine e dal complemento19. Analogamente a quanto osservato anche nel caso clinico descritto, è possibile osservare in questi pazienti un forte consumo complementare ascrivibile alla deposizione di frazioni del complemento a livello dei capillari glomerulari, oltre che alla deposizione anche delle IgM stesse nel medesimo sito che amplificano l’attivazione della cascata complementare.
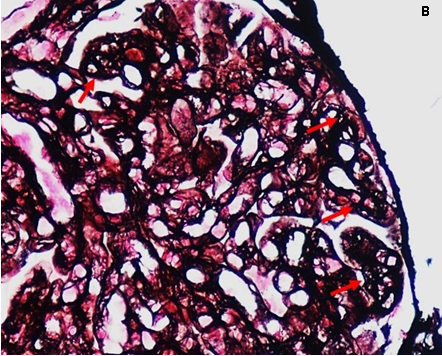
Figura 1: B) doppi contorni delle membrane basali dei capillari glomerulari con interposizione cellulare (frecce rosse). (Colorazione argento di Jones)
L’approccio terapeutico nel paziente con coinvolgimento renale in corso di disordine linfoproliferativo si basa sul trattamento della patologia ematologica di base caratterizzato nel caso clinico descritto, dall’associazione di Rituximab, Ciclofosfamide e Desametasone dimostrato notevole efficacia con contestuale basso profilo di tossicità20. L’efficacia della plasmaferesi nella rimozione delle paraproteine e conseguentemente nel miglioramento dell’outcome renale in presenza di un danno glomerulare associato a crioglobulinemia deve essere bilanciata con la necessità di trattamento della patologia linfoproliferativa con rituximab21. Infatti se l’utilizzo di rituximab, anticorpo monoclonale anti-linfociti B CD20+, consentirebbe di ridurre drasticamente il clone linfo-plasmacelluare e conseguentemente la produzione di paraproteine, l’applicazione di un trattamento plasmaferetico a distanza ravvicinata dalla somministrazione del rituximab rimuoverebbe una percentuale significativa di farmaco riducendone l’efficacia. Pertanto, il timing della somministrazione di rituximab deve essere valutato attentamente sulla base della severità malattia linfoproliferativa tenendo in considerazione che le terapie aferetiche rimangono una valida opzione terapeutica in particolare nelle seguenti situazioni cliniche: i) inefficacia della chemioterapia, ii) comparsa di segni clinici riferibili a tossicità o eventi avversi della chemioterapia e iii) severità del coinvolgimento di organi quali rene, SNC, cuore etc in corso di concomitante crioglobulinemia. Le crioglobulinemie che come è noto possono associarsi a disordini linfoproliferativi si associano a gradi variabili di vasculite sistemica, danno renale e neurologico le manifestazioni cliniche vanno da forme molto modeste a forme con complicanze renali, neuropatie e vasculiti sistemiche. La patologia è il risultato della precipitazione, nei piccoli vasi, di immunoglobuline o dei loro complessi, che inducono, poi, il processo di attivazione dei componenti del sistema complementare e la migrazione leucocitaria con danno tissutale; in tale contesto è stato in più occasioni dimostrata l’efficacia dei trattamenti aferetici quale supporto del trattamento farmacologico.
Nel caso clinico descritto l’utilizzo della plasmaferesi ha rappresentato il punto di svolta nel momento in cui è stato documentato un severo peggioramento funzionale renale con oligo-anuria, contestualmente al riscontro di un significativo incremento delle crioglobuline circolanti.
Nel paziente in questione è stato impiegato un plasma trattamento semi-selettivo con doppia filtrazione o filtrazione a cascata; tale trattamento impiega filtri plasmatici a diversa porosità per diverse sostanze, garantendo una maggiore selettività dei plasma-trattamenti rispetto al plasma-exchange soprattutto nella rimozione di macroglobuline tra cui immunoglobuline IgM, IgG, fibrinogeno, lipoproteine etc.
La filtrazione a cascata risparmia in modo efficace molte proteine autologhe, soprattutto l’albumina e alcuni fattori della coagulazione; inoltre non altera significativamente gli elettroliti, ad eccezione degli ioni divalenti che sono chelati dal citrato utilizzato per l’anticoagulazione.
Le metodiche di plasma filtrazione sono da considerare un’opportunità alternativa al plasma-exchange in caso il patogeno sia noto e si disponga di una membrana adatta alla molecola da sottrarre.
La filtrazione a cascata rispetto al plasma-exchange, è più selettiva, permette di processare più di un volume plasmatico ed è meno costosa a parità di efficacia clinica.
Un possibile svantaggio è rappresentato da un problema di biocompatibilità: se ne sconsiglia l’uso in pazienti che assumono ACE-inibitori per il rischio di reazioni anafilattoidi e di ipotensioni severe causate dal contatto del sangue con le membrane che causano l’inibizione delle kinine in pepetidi inattivi con conseguente rilascio di bradichinine.
Il coinvolgimento renale in corso di disordini linfoproliferativi può assumere diversi connotati, dando luogo a manifestazioni che sono l’esito del coinvolgimento, da parte delle paraproteine, delle diverse strutture renali. Sebbene le manifestazioni renali in corso di macroglobulinemia di Walden- strom siano meno frequenti rispet- to a quanto osservato in corso di altri disordini come ad esempio il mieloma multiplo, non va sot- tovalutato il possibile ruolo di tale patologia ematologica nel so- stenere processi patologici come la crioglobulinemia e con essa le alterazioni glomerulari associate alla glomerulonefrite membranoproliferativa. La diagnosi istologi- ca attraverso l’esecuzione di una biopsia renale nel caso di segni di danno d’organo (ematuria, protei- nuria e/o insufficienza renale) in corso di malattie linfoproliferative consente ottimizzare la terapia migliorando la prognosi a breve e a lungo termine del paziente.
Bibliografia
1. Sethi S, Fervenza FC, Zhang Y, et al. Proliferative glomerulonephritis secondary to dysfunction of the alter- native pathway of complement. Clin J Am Soc Nephrol 2011;6:1009-17
2. Sethi S, Fervenza FC. Membrano- proliferative glomerulonephritis: pa- thogenetic heterogeneity and proposal for a new classification. Semin Nephrol 2011;31: 341-8
3. Sethi S, Zand L, Leung N, et al. Membranoproliferative glomerulo- nephritis secondary to monoclonal gammopathy. Clin J Am Soc Nephrol 2010;5:770-82
4. Sethi S, Fervenza FC. Membranopro- liferative glomerulonephritis–a new look at an old entity. N Engl J Med. 2012 Mar 22;366(12):1119-31.
5. De Sanctis LB, Sestigiani E, Sgarlato V , Fabbrizio B, Santoro A. Il coinvol- gimento renale nelle gammopatie mo- noclonali e nel mieloma. G Ital Nefrol 2010; 27 (S50): S19-S33
6. Santostefano M, Zanchelli F, Zacca- ria A, Poletti G, Fusaroli M. The ultra- structural basis of renal pathology in monoclonal gammopathies. J Nephrol 2005; 18: 659-75
7. Merlini G, Pozzi C. Mechanisms of renal damage in plasma cell dyscra- sias: an overview. Contrib Nephrol 2007; 153: 66-86
8. Gertz MA; Waldenström macroglo- bulinemia: 2017 update on diagnosis, risk stratification, and management. Am J Hematol. 2017 Feb;92(2):209-217.
9. Kratochvil D, Kerstin Amann, Heike B, Maike B. Membranoproliferative glomerulonephritis complicating Wal- destrom’s Macroglobulinemia; BMC Nephrology 2012, 13:172.
10. Mutluay R, Aki SZ, Erten Y, Kon- ca C, Yagci M, Barit G, Sindel S: Mem- branoproliferative glomerulonephritis and light-chain nephropathy in asso- ciation with chronic lymphocytic leu- kemia. Clin Nephrol 70: 527-531, 2008
11. Higging L, Nars HS, Said SM, Kapoor P, Dingli D et al. Kindney in- volvement of patients with Walde- strom Macroglobulinemia and other IgM-producing B cell Lymphoproli- ferative disorders. Clin J Am Soc Ne- phrol 13:1037-1046, 2018
12. Chauvet S, Bridoux F, Ecotière L et al. Kidney disease associated with mo- noclonal Immunoglobuln M-secreting B-cell lymphoproliferative disorders: a case series of 35 patients. AM J Kidney Dis 2015
13. Vos JM, Gustine J, Rennke HG, Hunter Z et. Renal disease related to waldestrom macroglobulinemia; incidence, pathology and clinical outco- mes. British Journal of Haematology 2016, 175, 623-630.
14. Wechalekar AD, Lachmann HJ, Go- odman HJ. AL Amyloidosis associated with IgM paraproteinemia: clinical profile and treatment out come. Blood 2008; 112:4009-4016
15. Neel A, Perrin F, Decaux O et al. Long-term out come of monoclonal (type1) crioglobulinemia. Am J Hema- tol 2014; 89:156-161
16. Gnemmi V, Leleu X, Provot F. Cast Nephropathy and light chain deposi- tion disease in Waldestrom macroglo- bulinemia. Am J Kidney Dis 2012; 60: 487-491.
17. Miura N, Suzuki K, Yoshino M, Ki- tagawa W, Yamada H, Ohtani H, Joh K, Imai H: Acute renal failure due to IgMlambda glomerular thrombi and MPGN-like lesions in a patient with angioimmunoblastic T-cell lymphoma. Am J Kidney Dis 48: e3–e9, 2006
18. Ahad Lodhi, Abhishek Kumar, Muhammad U. Saqlain, and Manish Suneja. Thrombotic microangiopathy associated with proteasome inhibitors. Clinical Kidney Journal, 2015, vol. 8, no. 5, 632–636
19. Haraguchi S, Tomiyoshi Y, Aoki S, Sakemi T: Nephrotic syndrome due to immunologically mediated hypocom- plementic glomerulonephritis in a pa- tient of Waldenstrom’macroglobuline- mia. Nephron 92: 452–455, 2002
20. Dimopoulos MA, Anagnostopou- los A, Kyrtsonis MC, et al. Primary treatment of Waldenstrom macroglo- bulinemia with dexamethasone, ritu- ximab, and cyclophosphamide. J Clin Oncol 2007; 25:3344–3349.
21. Schwartz J, Padmanabhan A, Aqui N et al. Guidelines on the Use of The- rapeutic Apheresis in Clinical Practi- ce—Evidence-Based Approach from the Writing Committee of the Ameri- can Society for Apheresis: The Seventh Special Issue. Journal of Clinical Aphe- resis 2016; 31: 149-338.
22. P. Marson, G. Monti, F. Montani et al. Apheresis treatment of cryoglobuli- nemic vasculitis: A multicentre cohort study of 159 patients. Trasfusion and apheresis science, June 2018
23. Sanchez AP, Cunard R, Ward DM. The Selective Therapeutic Apheresis Procedures. Journal of Clinical Aphe- resis 2013; 28:20-29.
24. Mariano M. Tecniche di Aferesi. Giornale Italiano di Nefrologia 2017 Sep 28;34(5):73-88
25. Moreau ME, Adam A. Multifacto- rial aspect of acute side effects of an- giotensin converting enzyme inhibi- tors. Ann Pharm Fr 2006; 64:279-86