Devis Benfaremo, Emanuele Filippini, Armando Gabrielli



Dipartimento di Scienze Cliniche e Molecolari, Sezione di Clinica medica
Facoltà di Medicina e Chirurgia, Università Politecnica delle Marche
Scenario clinico
Un giovane di 23 anni, originario della Tunisia, viene ricoverato per l’inquadramento diagnostico-terapeutico di un quadro clinico caratterizzato da recente riscontro di pancitopenia (WBC 3350/mmc, Hb 9.6 g/dl, PLT 97.000/mmc) in paziente che riferisce comparsa da circa un anno di artralgie polidistrettuali, prevalentemente a carico di caviglie, ginocchia e polsi bilateralmente, e pregresse lesioni cutanee a carico di piedi e gomiti.
Approfondendo l’anamnesi, è emerso che il dolore ha coinvolto inizialmente entrambe le caviglie, con episodi di tumefazione articolare e rigidità mattutina della durata complessiva di circa un’ora. Successivamente, il dolore e la tumefazione hanno coinvolto le ginocchia, con parziale beneficio a seguito di assunzione di analgesici da banco. Dopo qualche mese venivano coinvolti anche cingolo scapolare e mani-polsi, e si assisteva a comparsa di lesioni bollose non meglio precisate a carico di piedi e gomiti, regredite spontaneamente. In Tunisia i reperti clinici venivano inquadrati con il generico termine di “reumatismo”.
Rientrato in Italia, il paziente si sottoponeva ad esami radiologici su suggerimento del medico curante. Eseguiva pertanto una risonanza magnetica (RM) del bacino con riscontro di alterazione puntiforme di segnale a carico di entrambe le articolazioni sacroiliache, compatibile con cisti subcondrale ma non con sacroileite; una radiografia (Rx) di mani-polsi, che risultava nei limiti di norma; e una Rx della colonna in toto, che mostrava unicamente una lieve rotoscoliosi dorsale destro-convessa.
L’anamnesi patologica remota è sostanzialmente muta, eccezion fatta per un pregresso intervento di appendicectomia ed un riscontro di lieve piastrinopenia fin dalla tenera età non sottoposta ad ulteriori approfondimenti.
Obiettivamente, il medico di reparto apprezza delle linfoadenomegalie sovraclaveari e inguinali bilaterali, non dolenti e di consistenza teso-elastica, mentre non si osservano anomalie a livello cardio-toraco-addominale né alterazioni neurologiche focali. A livello cutaneo, è presente un eritema a farfalla in regione zigomatica, mentre non si rilevano segni di alopecia né afte oro-genitali. La visita viene completata con l’esame dell’apparato muscoloscheletrico, ove si reperta dolorabilità alla palpazione di tutte le articolazioni metacarpofalangee (MCF) di entrambe le mani, in assenza di franchi segni di sinovite.
Dopo aver raccolto i dati clinico-anamnestici ed eseguito l’esame fisico, il medico di reparto definisce così il problema clinico:
Riscontro di pancitopenia in paziente con episodi di artrite simmetrica, flussionaria e migrante e rash eritematoso al volto.
A questo punto, prima di iniziare un percorso diagnostico, il medico si chiede se i disturbi articolari, il rash cutaneo e le alterazioni di laboratorio possano essere ricondotti ad un’unica ipotesi diagnostica o se invece siano espressione di due o più problematiche distinte.
Sulla base dei dati raccolti, ritiene che le ipotesi diagnostiche più probabili siano
A questo punto il medico si pone diversi quesiti clinici.
Quesiti diagnostici: quali sono gli esami da richiedere nel sospetto di una malattia autoimmune sistemica? Quali sono i criteri che ci permettono di porre la diagnosi di LES?
Malattia autoimmune sistemica, soprattutto il lupus eritematoso sistemico (LES) o una infezione virale; meno probabile il disturbo linfoproliferativo o una sindrome paraneoplastica.
La dimostrazione della presenza di autoanticorpi (in particolare, degli anticorpi anti-nucleo, ANA) è di solito il primo passo nella diagnosi di una malattia autoimmune sistemica, sebbene non sia sufficiente. L’eterogeneità delle manifestazioni cliniche delle malattie autoimmuni, infatti, fa sì che la diagnosi debba essere basata su un fondato sospetto clinico.
La diagnosi di LES si basa pertanto sul sospetto clinico del medico. Tuttavia, i criteri classificativi delle Società Scientifiche internazionali vengono spesso utilizzati a scopo diagnostico. I criteri più recenti sono stati appena pubblicati dall’American College of Rheumatology insieme all’European League Against Rheumatism (ACR/EULAR) nel 2019 (Tab. 1). Nel nostro paziente sono stati applicati i precedenti criteri SLICC/ACR del 2012 (Tab. 2), che risultano comparabili in termini di sensibilità e specificità.
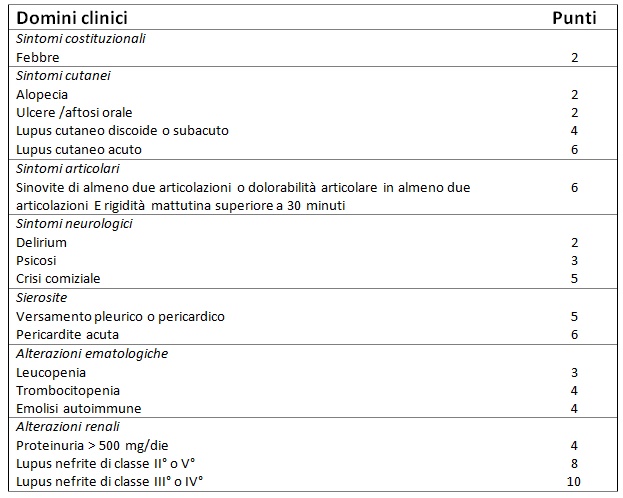
Tabella 1 – Nuovi criteri classificativi ACR/EULAR 2019 Tutti i pazienti classificati come affetti da LES devono avere anticorpi antinucleo (ANA) a livello siero ad un titolo di almeno 1:80 (entry criteria), oltre che totalizzare un punteggio superiore a 10 tra i criteri sopra riportati
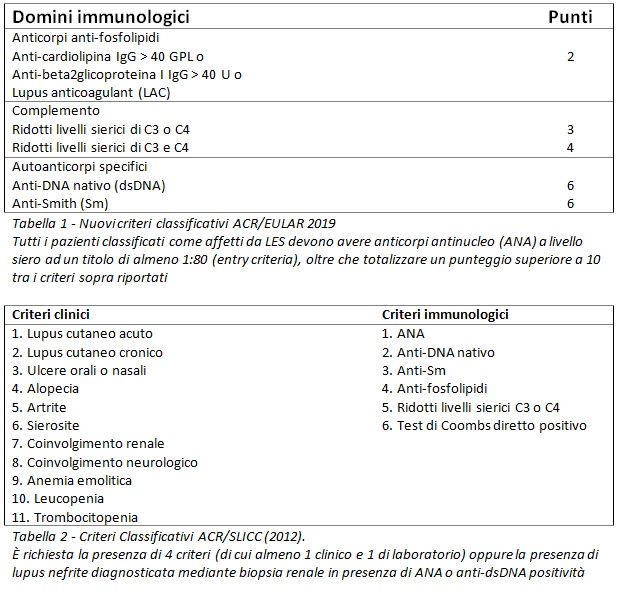
La dimostrazione della presenza di autoanticorpi (in particolare, degli anticorpi anti-nucleo, ANA) è di solito il primo passo nella diagnosi di una malattia autoimmune sistemica, sebbene non sia sufficiente. L’eterogeneità delle manifestazioni cliniche delle malattie autoimmuni, infatti, fa sì che la diagnosi debba essere basata su un fondato sospetto clinico.
La diagnosi di LES si basa pertanto sul sospetto clinico del medico. Tuttavia, i criteri classificativi delle Società Scientifiche internazionali vengono spesso utilizzati a scopo diagnostico. I criteri più recenti sono stati appena pubblicati dall’American College of Rheumatology insieme all’European League Against Rheumatism (ACR/EULAR) nel 2019 (Tab. 1). Nel nostro paziente sono stati applicati i precedenti criteri SLICC/ACR del 2012 (Tab. 2), che risultano comparabili in termini di sensibilità e specificità.
Per verificare le ipotesi diagnostiche, il medico di reparto richiede i seguenti esami: emocromo, che mostra leucopenia (leucociti 3350/mmc, 76% di neutrofili e 17% di linfociti), anemia normocitica (emoglobina 9.6 g/dl, MCV 92 fl), piastrinopenia (97.000/mmc), indici di flogosi (VES 54 mm/h, PCR 1.6 mg/dl, ferritina 397 ng/dl), frazioni del complemento, che risultano consumate (C3 64 mg/dl, C4 6 mg/dl), proteinuria delle 24 ore (500 mg nelle 24 ore, 150 mg nelle 24 ore ad un secondo controllo) e pannello autoimmunità (anticorpi anti-DNA nativo positivi ad alto titolo, LAC positivo, IgG e IgM anti-cardiolipina positivi, IgG e IgM anti Beta2glicoproteina I positivi, anti-Sm positivi, anti-U1RNP positivi).
Per escludere un disordine ematopoietico vengono inoltre richiesti: tipizzazione linfocitaria da sangue periferico che mostra riduzione consensuale delle linee linfocitarie; esame citologico da aspirato midollare il quale non evidenzia turbe della maturazione o accumulo di cellule immature; esame istologico su biopsia osteomidollare che reperta unicamente una lieve iperplasia delle tre serie ematopoietiche, maturanti.
Infine, per escludere un possibile disturbo eteroproliferativo viene eseguita una TC torace-addome, che evidenzia unicamente linfoadenomegalie di verosimile natura reattiva in sede ascellare e inguinale.
Pertanto, sulla base della presenza di eritema cutaneo, artrite, leucopenia, piastrinopenia, positività di ANA, anti-DNA nativo, anti-Sm, anti-fosfolipidi e consumo complementare, il nostro paziente soddisfa pienamente i suddetti criteri SLICC/ACR. Il medico pone quindi diagnosi di LES ad interessamento ematologico, cutaneo e articolare, ma si trova di fronte ad ulteriori quesiti.
Quesito diagnostico: in un paziente con LES, la biopsia renale è sempre necessaria? Quali sono le indicazioni e quali le controindicazioni all’esecuzione della stessa?
Un coinvolgimento renale clinico si verifica in circa il 60% dei pazienti con LES ed è una delle principali cause di morbilità e mortalità in questi pazienti. A meno che non vi sia una controindicazione, una biopsia renale dovrebbe essere eseguita nella maggior parte dei pazienti con lupus che presentano evidenza clinica o di laboratorio di coinvolgimento renale, al fine di ottenere una diagnosi corretta e la determinazione del sottotipo istologico di lupus nefrite.
Nello specifico, i criteri che rappresentano un’indicazione assoluta alla biopsia renale sono i seguenti:
– Proteinuria superiore a 500 mg/24 ore;
– Presenza di sedimento urinario attivo (ad es. ematuria e/o cilindri cellulari);
– Una creatinina sierica in aumento che non è chiaramente attribuibile a un altro meccanismo.
È improbabile che i pazienti con lupus con sedimento inattivo e meno di 500 mg/die di proteinuria presentino una LES nefrite proliferativa o membranosa, sebbene possano presentare una minima malattia proliferativa mesangiale, che non richiede un trattamento immunosoppressivo.
Nel nostro paziente, pertanto, si decide collegialmente con i colleghi nefrologi di soprassedere all’esecuzione della biopsia renale, iniziando quanto prima una terapia sistemica nel tentativo di migliorare il compenso ematologico e procedendo eventualmente alla biopsia sulla base dell’andamento clinico.
Il medico si trova quindi di fronte a nuovi quesiti terapeutici.
Quesiti terapeutici: in un paziente con diagnosi di LES a coinvolgimento ematologico, cutaneo, articolare, qual è la terapia iniziale più appropriata?
La triplice positività degli anticorpi anti-fosfolipidi, in assenza di pregressa o attuale storia di tromboembolismo venoso, giustifica una profilassi con anticoagulanti orali? Qual è la gestione più appropriata nel nostro paziente?
Per rispondere al primo quesito, sono state prese in esame le Linee Guida EULAR del 2019 (Fig. 1). In un paziente con diagnosi di LES non-renale, corticosteroidi ed drossiclorochina sono indicati in tutti i pazienti a prescindere dall’attività di malattia, che va presa in considerazione per eventuale terapia aggiuntiva, da instaurarsi in caso di malattia moderato-severa. I principali farmaci che possono essere usati per il controllo delle manifestazioni cliniche del LES sono il metotrexate, l’azatioprina, gli inibitori della calcineurina (as es. ciclosporina, tacrolimus), il micofenolato mofetile e il belimumab, l’unico farmaco con indicazione specifica per questa malattia autoimmune. Nei casi severi o refrattari, è possibile usare ciclofosfamide o rituximab.
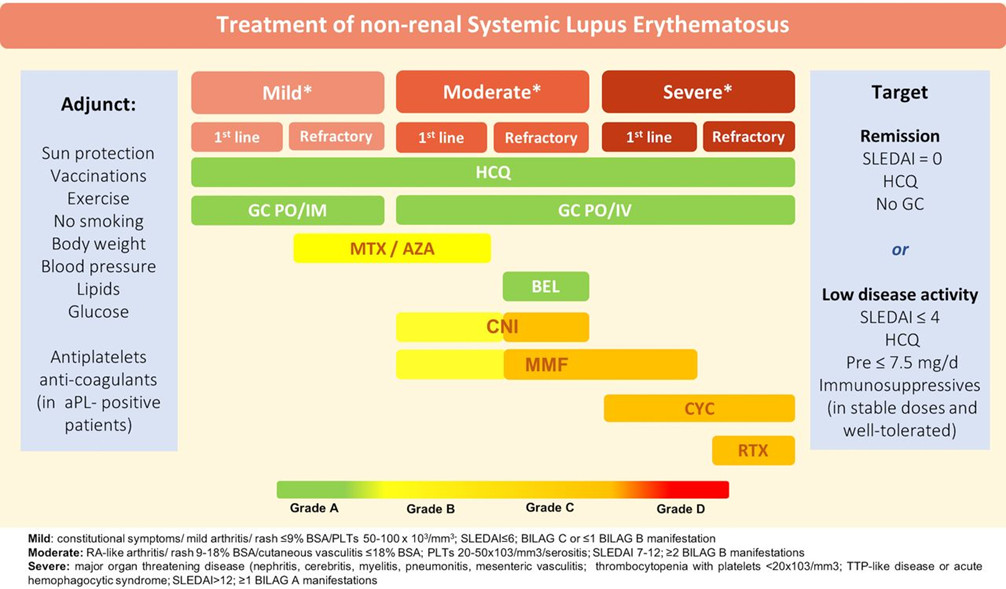
Figura 1 – Linee guida EULAR sul trattamento del LES non renale (2019)
La presenza di tripla positività degli anticorpi anti-fosfolipidi espone il paziente ad un alto rischio tromboembolico, tuttavia di per sé non è diagnostica di sindrome da anticorpi anti-fosfolipidi, che richiede in aggiunta la presenza di eventi tromboembolici. Misure generali da adottare in tutti i pazienti positivi includono lo screening e il controllo rigoroso dei fattori di rischio cardiovascolare (cessazione del fumo, gestione di ipertensione, dislipidemia e diabete, e regolare attività fisica) e prevenzione del tromboembolismo in situazioni ad alto rischio. Nei pazienti con LES e profilo anti-fosfolipidi ad alto rischio senza storia di trombosi o complicanze gravidiche, le linee guida raccomandano un trattamento profilattico con una bassa dose di aspirina. La terapia anticoagulante è indicata solo in prevenzione secondaria.
Nel nostro paziente si decide di intraprendere terapia con idrossiclorochina 5 mg/kg, prednisone 25 mg/die e profilassi con vitamina D, soprassedendo all’introduzione di antiaggreganti considerato il rischio emorragico secondario a trombocitopenia. Il paziente viene rivisto a controllo dopo circa 3 mesi, e alla luce della persistente piastrinopenia si decide di aggiungere ciclosporina 200 mg/die.
In seguito, il paziente viene perso al follow-up per motivi personali per circa due anni. Viene nuovamente ricoverato d’urgenza, dopo essersi sospeso autonomamente la terapia da circa un anno, con un quadro di polisierosite acuta ed insufficienza renale ingravescente (con proteinuria in range nefrosico), in seguito complicato da insufficienza respiratoria acuta da danno alveolare diffuso e stato anasarcatico che hanno richiesto terapia steroidea ad alte dosi, terapia antibiotica empirica, terapia diuretica endovenosa, cicli di ventilazione non invasiva ed emodialisi.
Nel corso di un successivo ricovero per nuovo peggioramento degli edemi declivi, all’esame ecocardiografico emerge una insufficienza aortica severa, con importante dilatazione delle camere cardiache ed immagini compatibili con vegetazioni endocarditiche a carico della valvola aortica. Il quadro viene confermato all’ecocardiografia transesofagea.
A questo punto il medico si trova di fronte a nuovi quesiti.
Quesiti terapeutici: quali sono gli elementi per sospettare una endocardite trombotica non-batterica? Qual è la terapia medica più appropriata? Quali sono le indicazioni al trattamento chirurgico?
L’endocardite trombotica non batterica o endocardite di Libman-Sacks è una complicanza rara ma temibile sia del LES che della sindrome da anticorpi anti-fosfolipidi. Rispetto alla forma infettiva, infatti, vi è una maggiore tendenza delle vegetazioni sterili ad embolizzare e causare infarti in diversi organi e apparati, che possono essere silenti o clinicamente manifesti. La diagnosi di endocardite di Libman-Sacks è difficile e si basa su un forte sospetto clinico nel contesto di malattie associate, come appunto il LES, la presenza di nuovi soffi cardiaci, la presenza di vegetazioni che non rispondono al trattamento antibiotico e/o l’evidenza di embolia sistemici multipla. Il trattamento medico di solito consiste in terapia anticoagulante sistemica in aggiunta al trattamento della malattia di base. Le linee guida ESC suggeriscono che l’indicazione all’intervento chirurgico sia la stessa dell’endocardite infettiva (ad es. insufficienza cardiaca, rottura acuta di valvola), sebbene la prevenzione di embolia ricorrente sia quella più comunemente riportata in letteratura.
Nel nostro paziente, vista la persistente negatività delle emocolture e l’assenza di risposta alla terapia antibiotica empirica, il sospetto clinico di endocardite trombotica non-batterica è fondato. Considerato il severo coinvolgimento della valvola aortica con insufficienza cardiaca, il paziente viene dunque trasferito in Cardiochirurgia, dove esegue impianto di protesi valvolare aortica di tipo meccanico, con decorso post-operatorio regolare.
Successivamente, per il persistere di attività clinica e laboratoristica di malattia, viene iniziata terapia con micofenolato mofetile, soprassedendo all’esecuzione della biopsia renale per l’elevato rischio emorragico.
Parole chiave: lupus eritematoso sistemico; anticorpi anti-fosfolipidi; endocardite trombotica non batterica
Letture consigliate
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019 Sep;78(9):1151-1159
- Hahn BH, McMahon MA, Wilkinson A, et al; American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken). 2012 Jun;64(6):797-808
- Fanouriakis A, Kostopoulou M, Alunno A, et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Annals of the Rheumatic Diseases 2019;78:736-745
- Tektonidou MG, Andreoli L, Limper M, et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann Rheum Dis. 2019 Oct;78(10):1296-1304
- Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC Guidelines for the management of infective endocarditis. Eur Heart J. 2015 Nov 21;36(44):3075-3128